Open AccessPosted Content
PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions.
TL;DR: A physics-informing strategy is proposed to predict the atom–atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein–ligand complex as their sum.
read more
Abstract: Recently, deep neural network (DNN)-based drug-target interaction (DTI) models are highlighted for their high accuracy with affordable computational costs. Yet, the models' insufficient generalization remains a challenging problem in the practice of in-silico drug discovery. We propose two key strategies to enhance generalization in the DTI model. The first one is to integrate physical models into DNN models. Our model, PIGNet, predicts the atom-atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein-ligand complex as their sum. We further improved the model generalization by augmenting a wider range of binding poses and ligands to training data. PIGNet achieved a significant improvement in docking success rate, screening enhancement factor, and screening success rate by up to 2.01, 10.78, 14.0 times, respectively, compared to the previous DNN models. The physics-informed model also enables the interpretation of predicted binding affinities by visualizing the energy contribution of ligand substructures, providing insights for ligand optimization. Finally, we devised the uncertainty estimator of our model's prediction to qualify the outcomes and reduce the false positive rates.
read more
Chat with Paper
AI Agents for this Paper
Find similar papers on Google Scholar, PubMed and Arxiv
Write a critical review of this paper
Analyze citations of this paper to find unaddressed research gaps
Figures

Fig. 2 The training scheme of PIGNet. We use three types of data in model training - true binding complex, true binder ligand-protein pair in computer-generated binding pose, and non-binding decoy complex. PIGNet predicts binding free energy for each input. For a true binding complex, the model learns to predict its true binding energy. The model also learns to predict the energy of a computergenerated binding pose complex or a non-binding decoy complex in higher value than the true binding energy and threshold energy, respectively. Finally, PIGNet learns the proper correlation of ligand atom position and binding affinity by minimizing the derivative loss. 
Fig. 3 Interpretation of the predicted outcomes. a. Substructural analysis of ligands for two target proteins. Protein-tyrosine phosphatase non-receptor type 1 (PTPN1) and platelet activating factor acetylhydrolase (PAF-AH). The blue and red circles indicate common and different substructures, respectively, and the predicted energy contribution (unit: kcal/mol) of each substructure is annotated. The inhibitory constant, Ki, indicates how potent the ligand binds to the target protein. b. A distance-energy plot of carbon-carbon pairwise van der Waals (VDW) energy components in the test set. The red solid line illustrates the original distance-energy relation without any deviation induced by learnable parameters. The closer the color of a data point to yellow, the larger the number of corresponding carbon-carbon pairs. c. The average value of the corrected sum of VDW radii, d′i j, corresponding to different carbon-carbon pair types. Csp2 −Csp2 , Csp2 −Csp3 , and Csp3 −Csp3 pairs are compared. The results include 95% confidence intervals. 
Table 2 The CASF-2016 benchmark results for the 3D GNN-based model and PIGNet (Single) with and without using data augmentation. The highest values within the same model are shown in bold 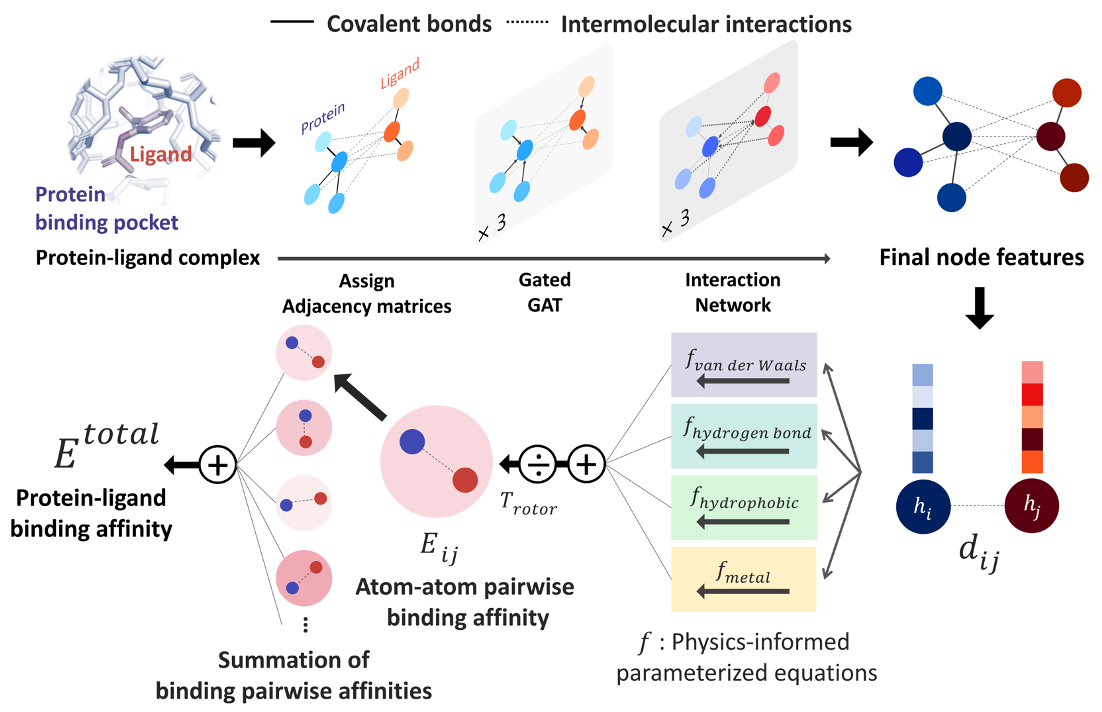
Fig. 1 Our model architecture. A protein-ligand complex is represented in a graph and adjacency matrices are assigned from the binding structure of the complex. Each node feature is updated through neural networks to carry the information of covalent bonds and intermolecular interactions. Given the distance and final node features of each atom pair, four energy components are calculated from the physics-informed parameterized equations. The total binding affinity is obtained as a sum of pairwise binding affinities, which is a sum of the four energy components divided by an entropy term. 
Fig. 4 Plot of the average Pearson’s correlation coefficients, R, of the 5-fold PIGNet model, with or without the uncertainty estimator, on the datasets classified according to the total uncertainty. PIGNet with the uncertainty estimator - low: the lowest third, random: the randomly selected one third, high: the highest third of the uncertainty distribution. PIGNet without Monte Carlo dropout - baseline: The scores of a single PIGNet model shown in the table 1. Error bars represent 95% confidence intervals. PIGNet was tested at the 2,300th training epoch with and without Monte Carlo dropout. 
Table 1 Benchmark test results on the CASF-2016 and the CSAR NRC-HiQ dataset. R, ρ indicate Pearson correlation coefficient and Spearman’s rank correlation coefficient, respectively. Top 1 score was used for a docking success rate, and top 1% rate was used for an average EF and a screening success rate. ∆VinaRF20 63 was excluded from the comparison, as it was fine-tuned on the PDBbind 2017 data, which in fact includes ∼ 50% of data in the CASF-2016 test set. The highest values of each column are shown in bold






Citations
DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Gabriele Corso,Hannes Stärk,Bowen Jing,Regina Barzilay,Tommi S. Jaakkola +4 more
- 04 Oct 2022
TL;DR: DiffDock as mentioned in this paper proposes a diffusion generative model over the non-Euclidean manifold of ligand poses to predict the binding structure of a small molecule ligand to a protein.
TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction
TL;DR: This paper proposes Trigonometry-Aware Neural networKs for binding structure prediction, TANKBind, that builds trigonometry constraint as a vigorous inductive bias into the model and explicitly attends to all possible binding sites for each protein by segmenting the whole protein into functional blocks.
Boosting Protein-Ligand Binding Pose Prediction and Virtual Screening Based on Residue-Atom Distance Likelihood Potential and Graph Transformer.
Chao Shen,Xujun Zhang,Yafeng Deng,Junbo Gao,Dong Wang,Lei-Jun Xu,Peichen Pan,Tingjun Hou,Yu Kang +8 more
TL;DR: In this article , a novel scoring function named RTMScore was developed by introducing a tailored residue-based graph representation strategy and several graph transformer layers for the learning of protein and ligand representations, followed by a mixture density network to obtain residue-atom distance likelihood potential.
106
Scoring Functions for Protein-Ligand Binding Affinity Prediction Using Structure-based Deep Learning: A Review
TL;DR: This work reviews structure-based scoring functions for binding affinity prediction based on deep learning, focussing on different types of architectures, featurization strategies, data sets, methods for training and evaluation, and the role of explainable artificial intelligence in building useful models for real drug-discovery applications.
Structure-based drug design with geometric deep learning
TL;DR: Geometric deep learning, an emerging concept of neural-network-based machine learning, has been applied to macromolecular structures as mentioned in this paper , highlighting its potential for structure-based drug discovery and design.
82
References
AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading
Oleg Trott,Arthur J. Olson +1 more
TL;DR: AutoDock Vina achieves an approximately two orders of magnitude speed‐up compared with the molecular docking software previously developed in the lab, while also significantly improving the accuracy of the binding mode predictions, judging by tests on the training set used in AutoDock 4 development.
•Posted Content
Empirical evaluation of gated recurrent neural networks on sequence modeling
TL;DR: These advanced recurrent units that implement a gating mechanism, such as a long short-term memory (LSTM) unit and a recently proposed gated recurrent unit (GRU), are found to be comparable to LSTM.
14.1K
Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences
Weizhong Li,Adam Godzik +1 more
TL;DR: Cd-hit-2d compares two protein datasets and reports similar matches between them; cd- Hit-est clusters a DNA/RNA sequence database and cd- hit-est-2D compares two nucleotide datasets.
10.7K
Cd-hit
TL;DR: A new CD-HIT program accelerated with a novel parallelization strategy and some other techniques to allow efficient clustering of such datasets to reduce sequence redundancy and improve the performance of other sequence analyses is developed.
Advances in Neural Information Processing Systems 14
08 Nov 2002
Abstract: The proceedings of the 2001 Neural Information Processing Systems (NIPS) Conference. The annual conference on Neural Information Processing Systems (NIPS) is the flagship conference on neural computation. The conference is interdisciplinary, with contributions in algorithms, learning theory, cognitive science, neuroscience, vision, speech and signal processing, reinforcement learning and control, implementations, and diverse applications. Only about 30 percent of the papers submitted are accepted for presentation at NIPS, so the quality is exceptionally high. These proceedings contain all of the papers that were presented at the 2001 conference. Bradford Books imprint
8.9K